IEC 62304:2021 is dead. Long live IEC 62304!

{fastsocialshare}
After not releasing the 2019 edition, the 2021 release was also officially cancelled in May this year.
This implies that IEC 62304:2006 + Amd1:2015 is still the most current and official version of the standard. Even if the last two attempts failed, this doesn’t mean that we cannot learn for the future. Sooner or later there will be a new edition of IEC 62304, just wait and see! Let's take a look at the proposed changes in the Version 2 that we will not be able to enjoy, given the cancellation.
New extended Scope
The older (IEC 62304:2006 + Amd1:2015) version was, as the title already implies, “Medical Device software – Software life-cycle processes, targeted for Software for Medical devices. The Version 2.0 title implied an enlarged scope to include any Health software, thus the proposed new name, ‘Health Software - Software life cycle processes’.
The scope boundaries are clarified by Figure 1:
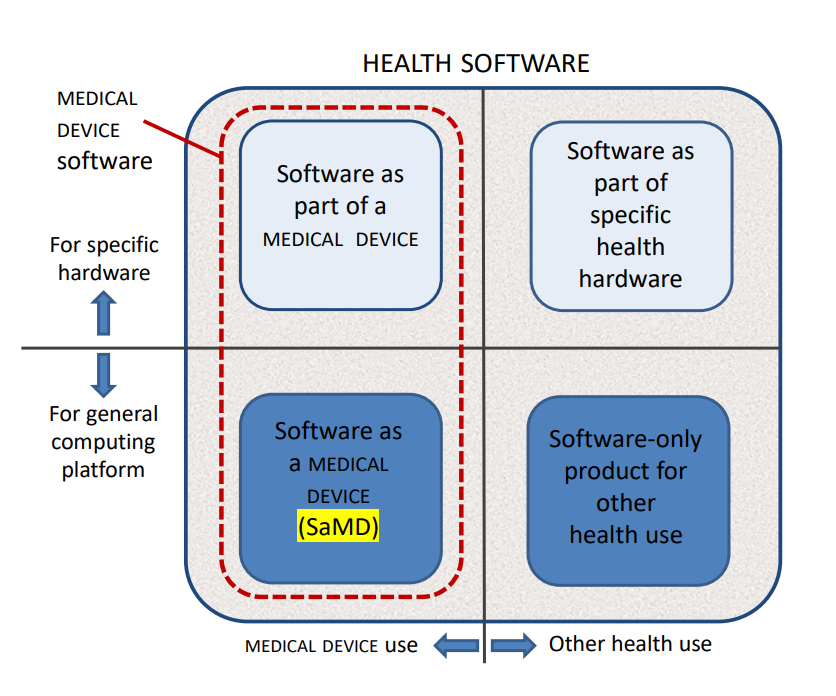 Figure 1 – HEALTH SOFTWARE field of application
Figure 1 – HEALTH SOFTWARE field of application
Any software-only medical device, is still regulated by IEC 82304-1:2016, but the whole sector of health devices would then have been covered by IEC 62304 Version 2.
Some examples to these categories:
- Software as a part of a medical device: software that is an integral part of a device, such as an infusion pump or dialysis machine.
- Software as part of specific health hardware: patient wristband software, healthcare software, health app on specific wearable hardware (i.e. watch, wristband, chest band).
- Software as a medical device (SaMD): software that is itself a medical device, such as a software application that performs diagnostic image analysis for making treatment decisions.
- Software-only product for other health use: hospital information systems, electronic health records, electronic medical records, mobile applications running on devices without physiologic sensors or detectors, software as a service, i.e. software executed in an external environment, providing calculation-results that fulfil the definition of a medical device.
Software Safety Class is now Software Process Rigor Level
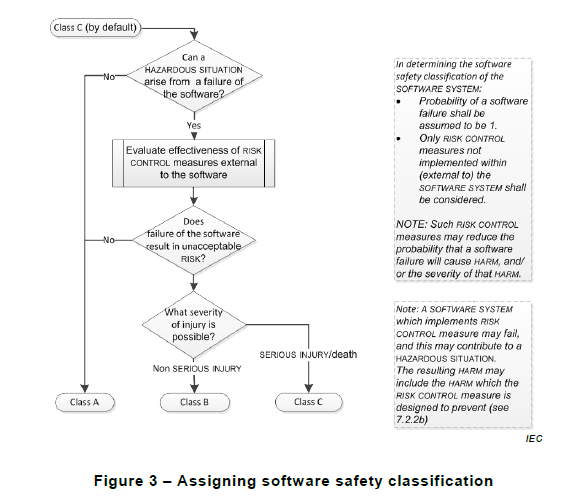
IEC 62304 has taught us to work with a Safety Classification for the Software system or parts of the Software system. In the proposed draft of Version 2, this would in the future change towards classifying the process needed to develop the Software system. The 2021 edition calls this Software process rigor level. This is intended to be the first activity in any software project. To perform a risk analysis on the software’s intended use worst-case scenarios to upfront detect and determine what process rigor to use.
The purpose of the software process rigour level is to determine the required rigour of the software processes prior to their start.
The classifications would still be:
A - Any software system failure cannot contribute to a hazardous situation leading to injury or death and considering external risk control measures, no risk controls are needed within the software system to reduce the risk of the hazardous situation.
B - A software system failure can contribute to a hazardous situation which results in non-serious injury.
C - When A or B does not apply. I.e. For serious injuries or death.
The proposed decision flow is described in Figure 2:
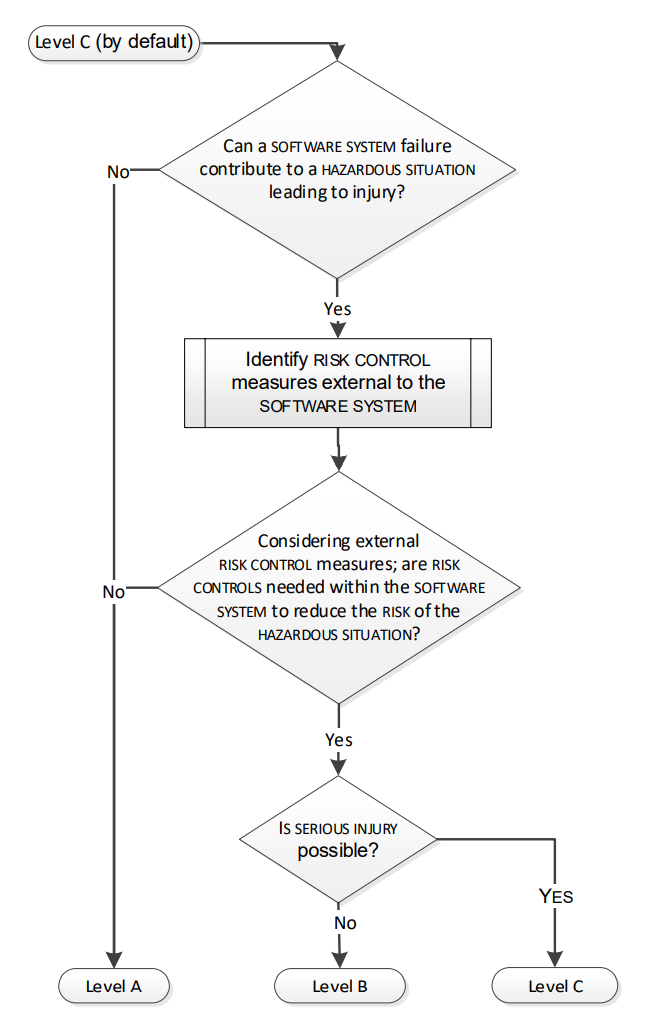
Figure 2 - Assigning software process rigour level
Now, the new approach has actually a big resemblance to how many companies have tried to tackle the challenge of finding a suitable Safety class. The most common approach that we have seen is that the complete system is labelled as A, B or C. Only rarely do we see different classifications used within the same development project. The classification system should in theory give us the possibility to gauge the documentation effort according to the risk posed by a particular part of the software, i.e. it is a system that should save us time and effort on the documentation level. So why not using it? The excuses we have comes across often lands in one of the following three categories:
- the effort of disentangling a software architecture that works great technically into an architecture that would isolate risky parts for "the sake of IEC 62304" is "not worthwhile"
- the effort of documenting the argumentation of why some parts are less risk is not worth the effort
- all parts of the software genuinely belongs to the same risk class
The possibility of using different rigour levels for different parts of the software system is still part of the proposed V2 draft, as are all the well-known decomposition levels ‘SW Item, SOUP, SW Unit and Detailed Design’. As we have got used to it from the 2006 + Amd1:2015 edition, you need to argue with some means of segregation between SW Item to assign different levels of rigour in the development process.
So why did IEC 62304:2021 fail?
How is it possible to work for so long on an update only to finally cancel its publishing? It is, honestly, the first time I ever hear of such a cancellation. How bad must the result have been to end up in the dustbin? For an in depth explanation, one of the committee members presents his view on the events that led to the cancellation in this article.
Mr. Kaestner later also published a poll on whether the software safety classifications should be deleted from the standard as a whole. A whopping 66% of the voters were happy to keep the Software Safety Classifications as they exist today, which is on the whole a proof that the community values a risk-based approach to medical device software development.






















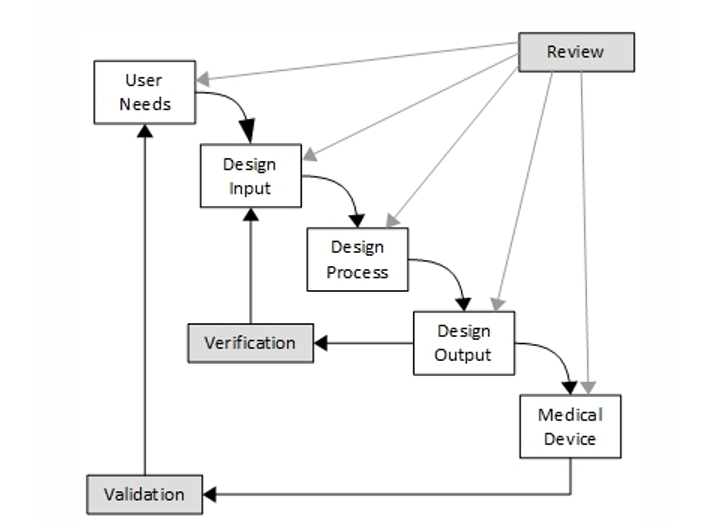







 It is rather rare that the developer has the real and deep experience a User has and it can therefore be precarious to leave the User Need elicitation process in the hands of an engineering team. Here are some tips for eliciting better User Needs.
It is rather rare that the developer has the real and deep experience a User has and it can therefore be precarious to leave the User Need elicitation process in the hands of an engineering team. Here are some tips for eliciting better User Needs.