5 Tips for Efficient Risk Assessments
Risk Assessments play a central role in Medical Device development. All medical device manufacturers apply risk management (they should because they have to!). All of them claim to be compliant with ISO 14971. And all of them do it differently.
I have worked with a large number of clients and I have seen more Risk Assessment variants than I can count. Some are good, some have, let's say, "potential".

From this experience, I can deduce a few best practices that will reduce the risk assessment effort considerably.
Here are my top five tips:
Don't brainstorm to identify risks
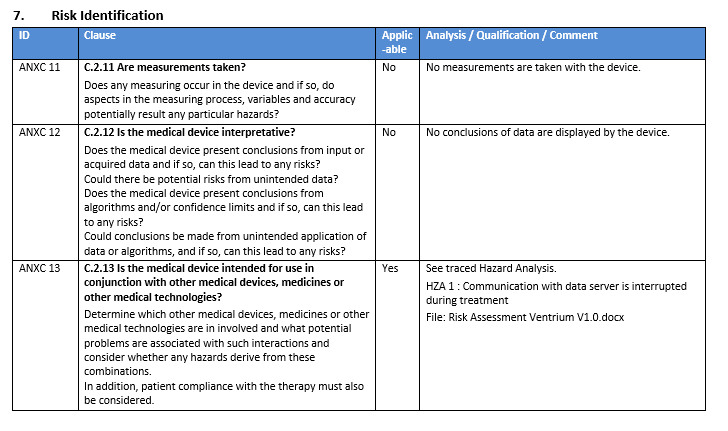
You are required to identify and assess ALL potential risks. How do you find them ALL? That can be a daunting question for someone new to the medical device industry.
However, the solution is to be structured i.e. to use a structured approach to systematically identify risks. There exist several known methods to do this, including:
- Task Analysis (analysing the use process)
- System Analysis (analysing the system through decomposition)
- Using the ISO 14971 annex questions
- Using existing risk reports of similar devices
Regardless of the approach selected, brainstorming should not be one of them. There are a number of well-known reasons for this, the most important one being that you will miss important risks.
Next time around, try a structured technique. You will identify more risks. I promise.
Use both top-down and bottom-up Risk Assessments
Some companies rely on EITHER bottom-up OR top-down risk assessment techniques and miss out on the fact that both approaches deliver vital and often DIFFERENT risks.
Top-down risk assessment techniques (such as PHA or Task Analysis) can be done early in the development process without much knowledge about the actual design of the device. It is a great tool for early identifying use errors and probably misuses.
Once the device design is known, the selected design itself must be analysed for risks (such as materials used, geometry, movements, and energy emittance, etc.) through a bottom-up risk assessment. FMEA's are very popular and well designed for this purpose. Both these techniques complement each other and should be conducted by any serious medical device manufacturer.
Don't keep Design Controls and Risk Management in separate systems
Design drives risk. And Risk drives design. This will become apparent when you need to follow up on the implementation and verification of mitigations as well as the further analysis if mitigations introduce new risks. The glue between the design and the risks is traceability. The effort of managing this traceability in a paper-based documentation system will be VERY high (those of you who have done it will nod now!).
So is applying software tools the solution? Not necessarily, since proper traceability monitoring can not be done until the requirement management tool is integrated with the risk management tool (or vice versa). Only by automatically managing the traceability between the Risk Assessment Items and the Design Items, preferably in a single tool, can true trace monitoring be obtained.
Use reasonable probability and severity scales
I am glad to see a clear trend of tightening down the probability and severity scales during the risk evaluations. From previously having used up to 10 steps, the current trend tends towards five to six steps or less. People simply have a very hard time judging whether a probability should be six or seven on a 1-10 scale and spend too much time pondering such questions. The range of options is simply too large to be effective!
For the probability axis, I would like to endorse Dr. Johner's approach of having each step representing 2 orders of magnitude. He explains this very well by saying, that apart from such an approach lets the probability axis span over more than 8 order of magnitudes, "...the factor 100 indicates the precision which we can appreciate... If you ask a group of people, how long it takes (on average) for a hard disk to be defective, the estimates vary between 2 years and 10 years. But everyone realizes that this average is greater than one month and less than 10 years. And between these two values is about a factor of 100."
Make use of existing mitigations
In many cases, the risk assessment is carried out when the design is already known. In such cases: when coming up with mitigations for your identified risks, use the already existing mitigations in your current design!
I bet your current design already contains a whole bunch of design decisions that are risk mitigations without you really considering them as such. The absolute majority of design teams I have encountered are very, very good at designing innovative and safe devices. However, many of the design decisions taken are based on previous experience, industry state-of-the-art, or simply old habits having been refined over time. Since these engineers are often better designers than document writers, they simply do not see their design (often already in place) through the lens of risk management.
Bottom line: your current design already contains of an uncovered treasure of existing mitigations. Try to use your existing design as mitigations when performing your next risk assessment.
Aligned Elements, our medical device ALM, assists you in performing structured risk assessments. Its highly customizable risk assessment configuration can be set up for a large array of risk analysis variants. Should you be interested in a demonstration, contact us at